
- Dirac Charge in Antiferromagnetic Topological Semimetals
-
Phys. Rev. Lett. 136, 046603 (2026)
Topological node of electronic bands can carry emergent charge degree of freedom such as the Berry curvature monopole of the Weyl semimetals, which results in intriguing transport and optical phenomena. In this Letter, we discuss the existence of the hidden “Dirac charge” and its detection via the photocurrent response in antiferromagnetic (AFM) Dirac semimetals. In light of the Berry curvature defined in the spin and spin-charge-mixed parameter space, we identify Dirac charges as sources or sinks of the Berry curvature in the generalized parameter space. We demonstrate that this Dirac charge can be detected via the photocurrent driven by the spin-charge-coupled motive force. By using real-time simulation, we find that the Dirac charge plays a significant role in the photocurrent generation in AFM Dirac semimetals. This Letter reveals the hidden property of the Dirac points in AFM Dirac semimetals.
- Calculation of the Biquadratic Spin Interactions Based on the Spin Cluster Expansion for Ab initio Tight-binding Models
-
J. Phys. Soc. Jpn 94, 124709 (2025) Editors' choice

We develop a calculation scheme using ab initio tight-binding Hamiltonians to evaluate biquadratic magnetic interactions. This approach relies on the spin cluster expansion combined with the disordered local moment (DLM) method, originally developed within the multiple scattering Korringa-Kohn-Rostoker method. Applying it to a single-orbital Hubbard model with two sublattices, we show that the evaluated DLM biquadratic interactions are in good agreement with those obtained from the strongly correlated limit, demonstrating the wide applicability of the method to various magnetic systems with large local moments. We then apply it to the ab initio tight-binding models for elemental magnetic metals; the resulting magnetic interactions align well with previous literature. Finally, we explore its performance in more complex compounds, such as transition metal dichalcogenides with intercalation of 3d transition metals and potassium electrosodalite. The obtained results for both compounds show good agreement with experiments. The present approach offers a convenient ab initio path for evaluating biquadratic interactions and understanding the electronic mechanisms controlling them.
- Symmetry analysis of cross-circular and parallel-circular Raman optical activity
-
Phys. Rev. B 112, 115105 (2025) Editors' suggestion
Raman scattering with regard to circularly polarized incident and scattered lights is closely related to the circular activity of a given system. We investigate the symmetry of its activity, called the cross-circular and parallel-circular Raman optical activity. The analysis is systematically performed with the magnetic point groups, and it indicates that the response allows for a useful diagnosis of the symmetry of materials such as chirality and (magneto)axiality. It is also shown that the Stokes and anti-Stokes processes are related to each other by the conserved antiunitary symmetry, which can be either the time-reversal operation or the combination of time-reversal and mirror reflection.
- Strong-coupling high-Tc superconductivity in doped correlated band insulators
-
Phys. Rev. B 112, L020504(2025)
We explore the superconducting properties of the bilayer Hubbard model, which exhibits a high transition temperature (Tc) for ans± pairing, using a cluster extension of the dynamical mean-field theory. Unlike the single-layer Hubbard model, where the d wave superconductivity emerges by doping the Mott insulator, the parent state of the bilayer system is a correlated band insulator. Above Tc, slight hole (electron) doping introduces a striking dichotomy between electron and hole pockets: The electron (hole) pocket develops a pseudogap while the other becomes a nearly incipient band. We reveal that the superconductivity is driven by kinetic (potential) energy gain in the underdoped (overdoped) region. We also find a very short coherence length, for which we argue the relevance to multiorbital physics. Our Letter offers crucial insights into the superconductivity in the bilayer Hubbard model potentially relevant to La3O2O7.
- Spin models and cluster multipole method: Application to kagome magnets
-
Phys. Rev. B 112, 014406(2025)

We present a multiscale computational approach that combines atomistic spin models with the cluster multipole (CMP) method. The CMP method enables a systematic and accurate generation of complex noncollinear magnetic structures using symmetry-adapted representations. The parameters of the spin model are derived from density functional theory using the magnetic force theorem, with the paramagnetic state as a reference. The energy landscape of CMP-generated structures is inspected at the model Hamiltonian level and sets of low-energy magnetic structures are identified for each material candidate. The inclusion of relativistic antisymmetric and anisotropic pair interactions lifts partially the degeneracy among these most stable structures. To demonstrate the applicability and predictive capability of the method, we apply it to the noncollinear Mn3X and collinear Fe3X (X=Ga,Ge, and Sn) kagome compounds. The computational efficiency of the method in identifying low-energy structures among multiple CMP configurations highlights its potential for high-throughput screening of complex magnets with unknown magnetic order.
- Multiferroic Collinear Antiferromagnets with Hidden Altermagnetic Spin Splitting
-
Phys. Rev. Lett. 134, 226703(2025)
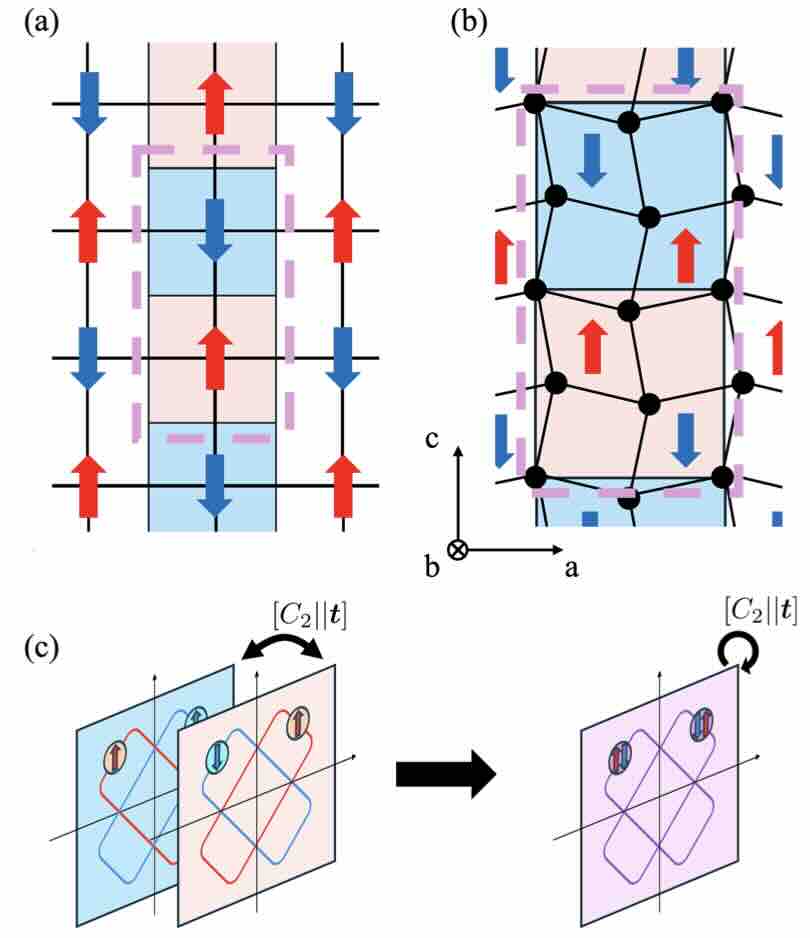
Altermagnets exhibit nonrelativistic spin splitting due to the breaking of time-reversal symmetry and have been garnering significant attention as promising materials for spintronic applications. In contrast, conventional antiferromagnets without spin splitting seem to not have any symmetry breaking and have drawn less attention. However, we show that conventional antiferromagnets with a nonzero Q vector bring about nontrivial symmetry breakings. The incompatibility between the Q vector and nonsymmorphic symmetry leads to macroscopic symmetry breaking without lifting spin degeneracy. Moreover, the hidden altermagnetic spin splitting in the electronic structure gives rise to various emergent responses. To examine our prediction, we perform first-principles calculations for MnS2 and investigate its multiferroic properties, such as nonlinear transport and optical activity. Our findings reveal unique properties in conventional antiferromagnets, providing another perspective for designing spintronic materials.
- Nonlinear Hall effect driven by spin-charge-coupled motive force
-
Phys. Rev. B 111, 174416(2025)
Parity-time-reversal symmetric (PT-symmetric) magnets have garnered much attention due to their spin-charge coupled dynamics enriched by parity-symmetry breaking. By real-time simulations, we study how localized spin dynamics can affect the nonlinear Hall effect in PT-symmetric magnets. To identify the leading-order term, we derive analytical expressions for the second-order optical response and classify the contributions by considering their transformation properties under PT symmetry. Notably, our results reveal that sizable contribution is attributed to the mixed dipole effect, which is analogous to the Berry curvature dipole term.
- Topological Hall Effect of Skyrmions from first Principles
-
Phys. Rev. X 15, 011054(2025)
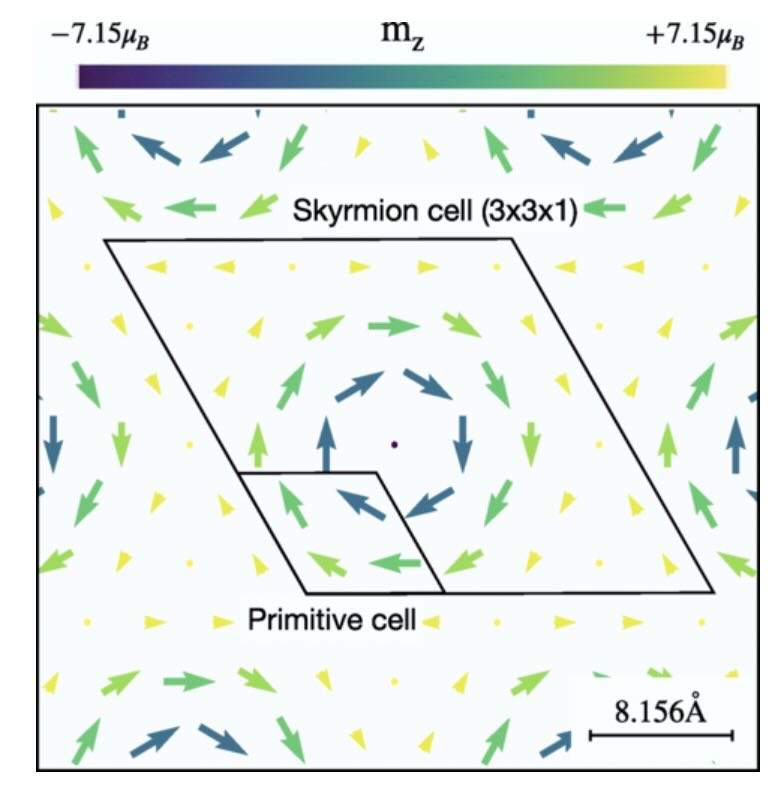
We formulate a first-principles approach for calculating the topological Hall effect (THE) in magnets with noncollinear nanoscale spin textures. We employ a modeling method to determine the effective magnetic field induced by the spin texture, thereby circumventing the computational challenges associated with superlattice calculations. Based on these results, we construct a Wannier tight-binding Hamiltonian to characterize the electronic states and calculate the Hall conductivity. Applying this approach to the skyrmion material Gd2PdSi3 shows good agreement with experimental data. Our analysis in momentum space further reveals that the dominant contribution to the THE arises from the crossing points between the folded bands along high-symmetry lines in the Brillouin zone. This work advances numerical techniques for simulating a general magnetic system, exemplified by but not restricted to skyrmion lattice, and its result offering insights into the complex interplay between spin textures and electronic transport.
- Ab initio study on magnetism suppression, anharmonicity, rattling mode, and superconductivity in Sc6M Te2 (M=Fe,Co,Ni)
-
Phys. Rev. B 110, 104505 (2024)
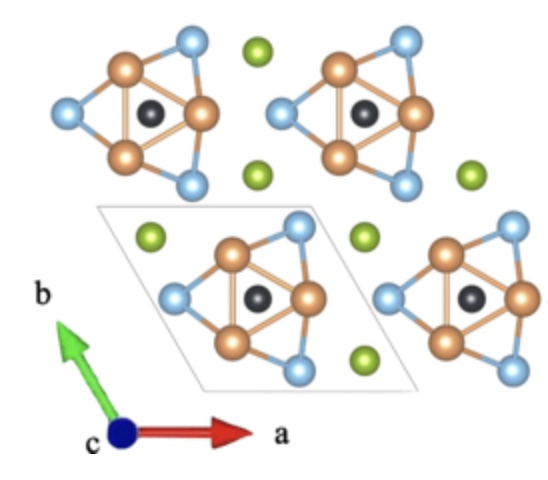
We perform a systematic ab initio study on phonon-mediated superconductivity in the transition-metal-based superconductors Sc6M Te2 (M=Fe,Co,Ni). Firstly, our charge analysis reveals significant electron transfer from Sc to because of the substantial difference in the electronegativity, filling the 3 d orbitals of M and suppressing magnetic instability. Secondly, we show that Sc6 FeTe2 exhibits strong lattice anharmonicity. Moreover, for M=Fe and Co, we find low-frequency soft phonon bands of M, which can be interpreted as rattling phonons in the framework formed by Sc. While not observed in the case of M=Ni, the rattling phonons give rise to a prominent peak or plateau in the Eliashberg spectral function and enhance the pairing instability. By reproducing the experimental trend of superconducting transition temperatures, our study underscores the potential of designing phonon-mediated superconductors by strategically combining nonsuperconducting and magnetic transition-metal elements.
- Continuous crossover between insulating ferroelectrics and polar metals: Ab initio calculation of structural phase transitions of LiBO3 (B=Ta, W, Re, Os)
-
Phys. Rev. B 110, 094102 (2024)
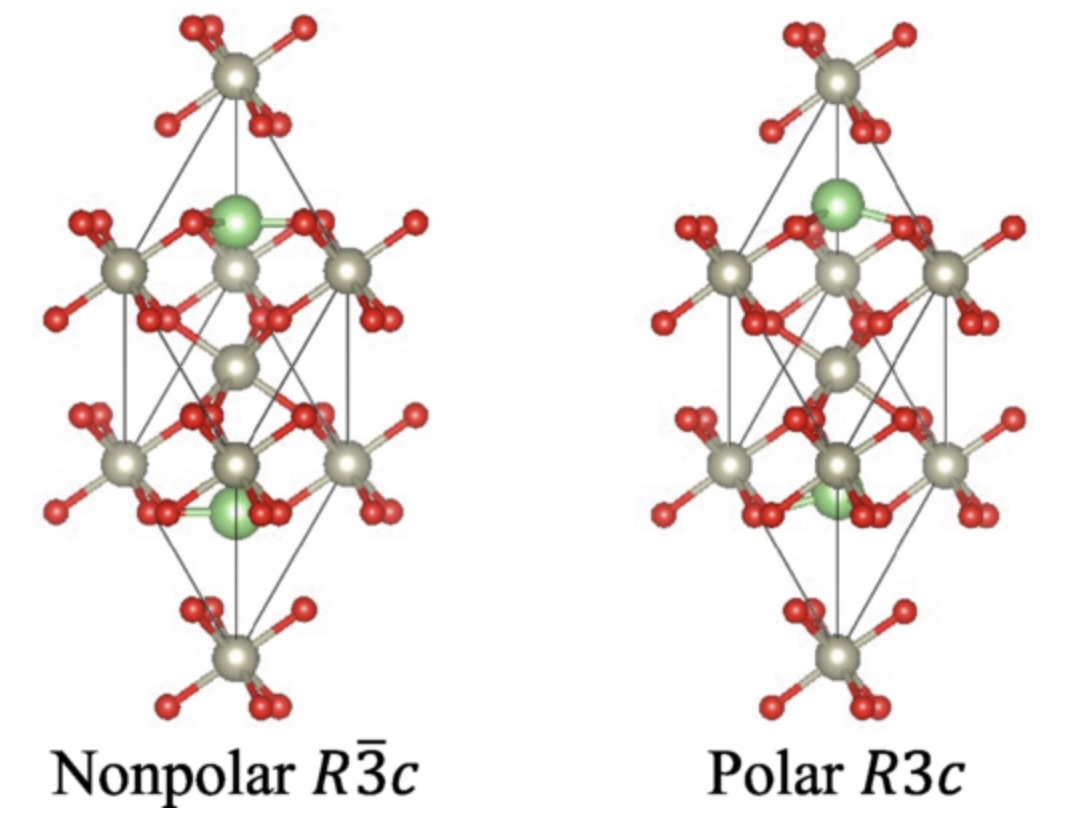
Inspired by the recent discovery of a new polar metal LiReO3 by K. Murayama et al, we calculate the temperature (T)-dependent crystal structures of LiBO3 with B=Ta, W, Re, Os, using the self-consistent phonon (SCPH) theory. We have reproduced the experimentally observed polar-nonpolar structural phase transitions and the transition temperatures (Tc) of LiTaO3, LiReO3, and LiOsO3. From the calculation, we predict that LiWO3 is a polar metal, which is yet to be tested experimentally. Upon doping electrons to the insulating LiTaO3, the predicted Tc is quickly suppressed and approaches those of the polar metals. Thus, there is a continuous crossover between ferroelectric insulators and polar metals if we dope electrons to the ferroelectric insulators. Investigating the detailed material dependence of the interatomic force constants (IFCs), we explicitly show that the suppression of Tc in polar metals can be ascribed to the screening of the long-range Li-O interaction, which is caused by the presence of the itinerant electrons. The quantitative finite-temperature calculations do not show signs of unscreened long-range interactions by the weak electron-phonon coupling or enhancement of polar instabilities by carrier doping, as expected in some previous works.
- First-principles study of the tunnel magnetoresistance effect with Cr-doped RuO2 electrode
-
Phys. Rev. B 110, 064433 (2024)
We investigate the functionality of the Cr-doped RuO2 as an electrode of the magnetic tunnel junction (MTJ), motivated by a recent experiment showing that Cr doping into rutile-type RuO2 is an effective tool to easily control its antiferromagnetic order and the resultant magnetotransport phenomena. We perform first-principles calculation of the tunnel magnetoresistance (TMR) effect in the MTJ based on the Cr-doped RuO2 electrodes. We find that a finite TMR effect appears in the MTJ originating from the momentum-dependent spin splitting in the electrodes, which suggests that RuO2 with Cr doping will function effectively as the electrode of the MTJ. We also show that this TMR effect can be qualitatively captured by the local density of states inside the tunnel barrier.
- Effect of collective spin excitations on electronic transport in topological spin textures
-
Phys. Rev. B 110, 014425 (2024)
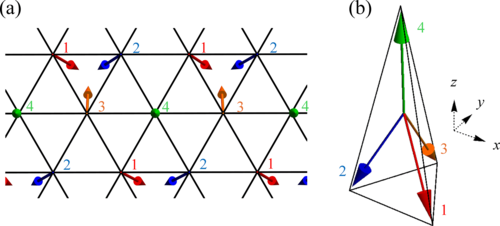
We develop an efficient real-time simulation method for the spin-charge coupled system in the velocity gauge. This method enables us to compute the real-time simulation for the two-dimensional system with a complex spin texture. We focus on the effect of the collective excitation of the localized spins on the electronic transport properties of the nontrivial topological state in real space. To investigate this effect, we calculate the linear optical conductivity by calculating the real-time evolution of the Kondo lattice model on the triangular lattice, which hosts an all-in/all-out (triple-Q) magnetic structure. In the linear conductivity spectra, we observe multiple peaks below the band gap regime, attributed to the resonant contributions of collective modes similar to the skyrmionic system, alongside broadband modifications resulting from off-resonant spin dynamics. This result shows that the collective excitation, similar to the skyrmionic system, influences the optical response of the electron systems based on symmetry analysis. We elucidate the interference between the contributions from the different spin excitations to the optical conductivity in the multiple-spin texture, pointing out the mode-dependent electrical activity. We show the complex interplay between the complex spin texture and the itinerant electrons in the two-dimensional spin-charge coupled system.
- Universal chemical formula dependence of ab initio low-energy effective Hamiltonian in single-layer carrier-doped cuprate superconductors: Study using a hierarchical dependence extraction algorithm
-
Phys. Rev. B 110, 014502 (2024)
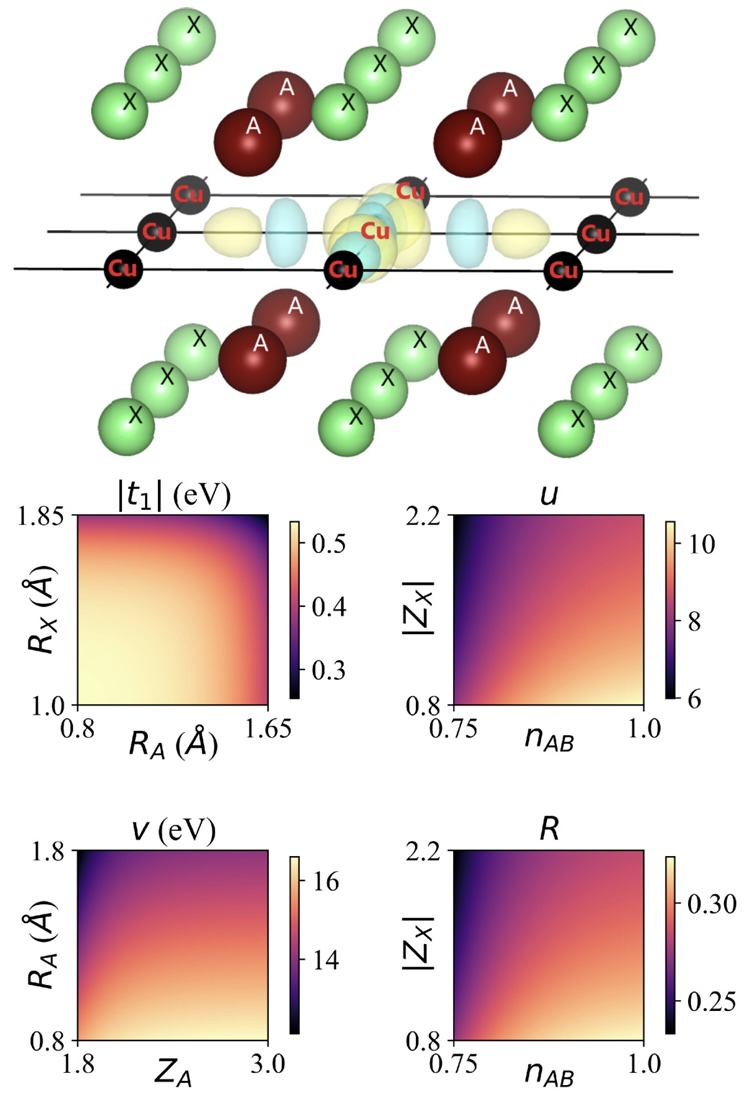
We explore the possibility to control the superconducting transition temperature at optimal hole doping Toptc in cuprates by tuning the chemical formula (CF). Toptc can be theoretically predicted from the parameters of the ab initio low-energy effective Hamiltonian with one antibonding (AB) Cu 3dx2-y2/O 2pσ orbital per Cu atom in the CuO2 plane, notably the nearest-neighbor hopping amplitude |t1| and the ratio u=U/|t1|, where U is the onsite effective Coulomb repulsion. However, the CF dependence of |t1| and u is a highly nontrivial question. In this paper, we propose the universal dependence of |t1| and u on the CF and structural features in hole doped cuprates with a single CuO2 layer sandwiched between block layers. To do so, we perform extensive ab initio calculations of |t1| and u and analyze the results by employing a machine-learning method called hierarchical dependence extraction (HDE). The main results are (a) |t1| has a main-order dependence on the radii RX and RA of the apical anion X and cation A in the block layer. (|t1| increases when RX or RA decreases.) (b) u has a main-order dependence on the ionic charge ZX of X and the hole doping δ of the AB orbital. (u decreases when |ZX| increases or δ increases.) We elucidate and discuss the microscopic mechanism of items (a) and (b). We demonstrate the predictive power of the HDE by showing the consistency between items (a) and (b) and results from previous works. The present results provide a basis for optimizing superconducting properties in cuprates and possibly akin materials. Also, the HDE method offers a general platform to identify dependencies between physical quantities.
- Symmetry analysis with spin crystallographic groups: Disentangling effects free of spin-orbit coupling in emergent electromagnetism
-
Phys. Rev. B 109, 094438 (2024) (Editors' suggestion)

Recent studies identified spin-order-driven phenomena such as spin-charge interconversion without relying on the relativistic spin-orbit interaction. Those physical properties can be prominent in systems containing light magnetic atoms due to sizable exchange splitting and may pave the way for realization of giant responses correlated with the spin degree of freedom. In this paper, we present a systematic symmetry analysis based on the spin crystallographic groups and identify the physical property of a vast number of magnetic materials up to 1500 in total. By decoupling the spin and orbital degrees of freedom, our analysis enables us to take a closer look into the relation between the dimensionality of spin structures and the resultant physical properties and to identify the spin and orbital contributions separately. In stark contrast to the established analysis with magnetic space groups, the spin crystallographic group manifests richer symmetry including spin-translation symmetry and leads to emergent responses. For representative examples, we discuss the geometrical nature of the anomalous Hall effect and magnetoelectric effect and classify the spin Hall effect arising from the nonrelativistic spin-charge coupling. Using the power of computational analysis, we apply our symmetry analysis to a wide range of magnets, encompassing complex magnets such as those with noncoplanar spin structures as well as collinear and coplanar magnets. We identify emergent multipoles relevant to physical responses and argue that our method provides a systematic tool for exploring sizable electromagnetic responses driven by spin order.
- High-throughput calculations of antiferromagnets hosting anomalous transport phenomena
-
Phys. Rev. B 109, 094435 (2024)
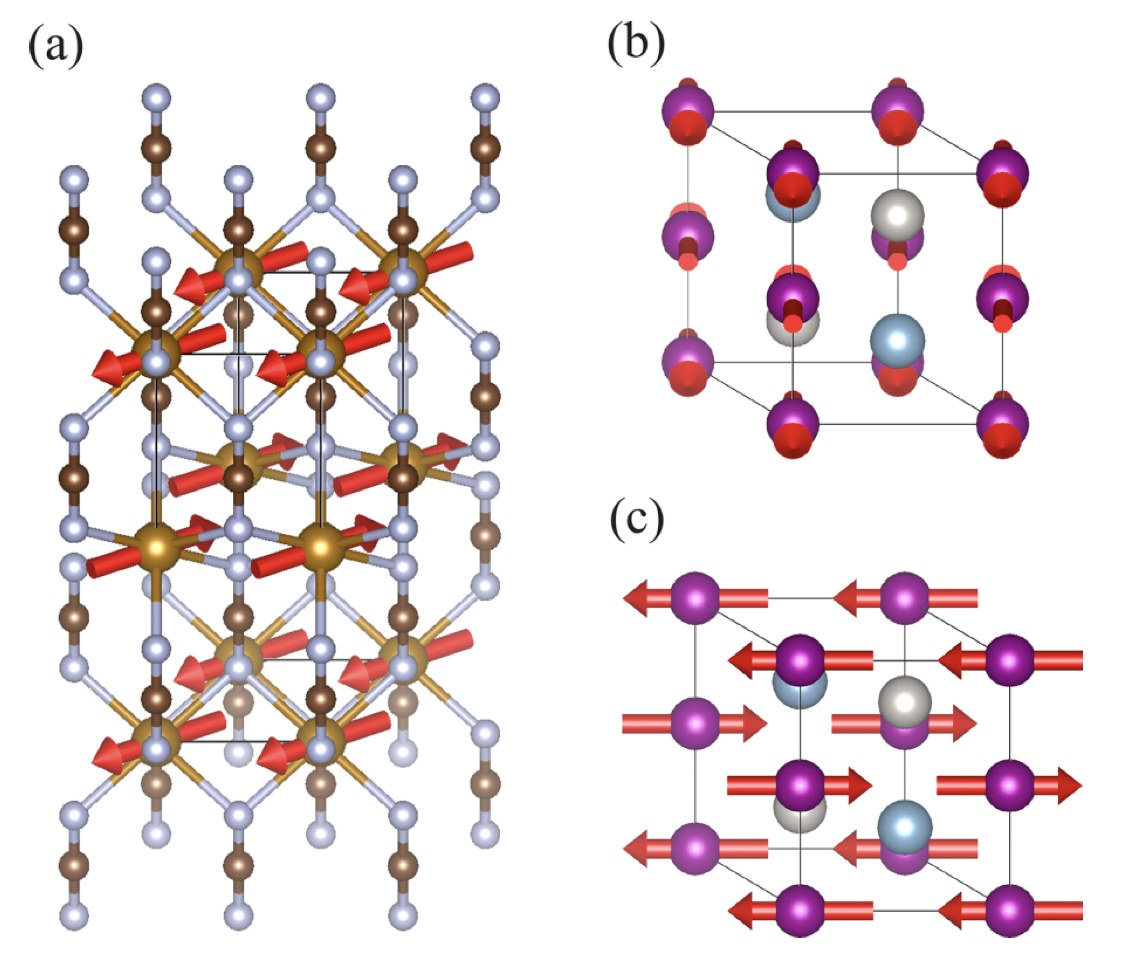
We develop a high-throughput computational scheme based on cluster multipole theory to identify new functional antiferromagnets (AFMs). This approach is applied to 228 magnetic compounds listed in the AtomWork-Adv database, known for their elevated Neel temperatures. We conduct systematic investigations of both stable and metastable magnetic configurations of these materials. Our findings reveal that 34 of these compounds exhibit AFM structures with zero propagation vectors and magnetic symmetries identical to conventional ferromagnets, rendering them potentially invaluable for spintronics applications. By cross-referencing our predictions with the existing MAGNDATA database and published literature, we verify the reliability of our findings for 26 out of 28 compounds with partially or fully elucidated magnetic structures in the experiments. These results not only affirm the reliability of our scheme but also point to its potential for broader applicability in the ongoing quest for the discovery of functional magnets.
 Topological node of electronic bands can carry emergent charge degree of freedom such as the Berry curvature monopole of the Weyl semimetals, which results in intriguing transport and optical phenomena. In this Letter, we discuss the existence of the hidden “Dirac charge” and its detection via the photocurrent response in antiferromagnetic (AFM) Dirac semimetals. In light of the Berry curvature defined in the spin and spin-charge-mixed parameter space, we identify Dirac charges as sources or sinks of the Berry curvature in the generalized parameter space. We demonstrate that this Dirac charge can be detected via the photocurrent driven by the spin-charge-coupled motive force. By using real-time simulation, we find that the Dirac charge plays a significant role in the photocurrent generation in AFM Dirac semimetals. This Letter reveals the hidden property of the Dirac points in AFM Dirac semimetals.
Topological node of electronic bands can carry emergent charge degree of freedom such as the Berry curvature monopole of the Weyl semimetals, which results in intriguing transport and optical phenomena. In this Letter, we discuss the existence of the hidden “Dirac charge” and its detection via the photocurrent response in antiferromagnetic (AFM) Dirac semimetals. In light of the Berry curvature defined in the spin and spin-charge-mixed parameter space, we identify Dirac charges as sources or sinks of the Berry curvature in the generalized parameter space. We demonstrate that this Dirac charge can be detected via the photocurrent driven by the spin-charge-coupled motive force. By using real-time simulation, we find that the Dirac charge plays a significant role in the photocurrent generation in AFM Dirac semimetals. This Letter reveals the hidden property of the Dirac points in AFM Dirac semimetals. Raman scattering with regard to circularly polarized incident and scattered lights is closely related to the circular activity of a given system. We investigate the symmetry of its activity, called the cross-circular and parallel-circular Raman optical activity. The analysis is systematically performed with the magnetic point groups, and it indicates that the response allows for a useful diagnosis of the symmetry of materials such as chirality and (magneto)axiality. It is also shown that the Stokes and anti-Stokes processes are related to each other by the conserved antiunitary symmetry, which can be either the time-reversal operation or the combination of time-reversal and mirror reflection.
Raman scattering with regard to circularly polarized incident and scattered lights is closely related to the circular activity of a given system. We investigate the symmetry of its activity, called the cross-circular and parallel-circular Raman optical activity. The analysis is systematically performed with the magnetic point groups, and it indicates that the response allows for a useful diagnosis of the symmetry of materials such as chirality and (magneto)axiality. It is also shown that the Stokes and anti-Stokes processes are related to each other by the conserved antiunitary symmetry, which can be either the time-reversal operation or the combination of time-reversal and mirror reflection. We explore the superconducting properties of the bilayer Hubbard model, which exhibits a high transition temperature (Tc) for ans± pairing, using a cluster extension of the dynamical mean-field theory. Unlike the single-layer Hubbard model, where the d wave superconductivity emerges by doping the Mott insulator, the parent state of the bilayer system is a correlated band insulator. Above Tc, slight hole (electron) doping introduces a striking dichotomy between electron and hole pockets: The electron (hole) pocket develops a pseudogap while the other becomes a nearly incipient band. We reveal that the superconductivity is driven by kinetic (potential) energy gain in the underdoped (overdoped) region. We also find a very short coherence length, for which we argue the relevance to multiorbital physics. Our Letter offers crucial insights into the superconductivity in the bilayer Hubbard model potentially relevant to La3O2O7.
We explore the superconducting properties of the bilayer Hubbard model, which exhibits a high transition temperature (Tc) for ans± pairing, using a cluster extension of the dynamical mean-field theory. Unlike the single-layer Hubbard model, where the d wave superconductivity emerges by doping the Mott insulator, the parent state of the bilayer system is a correlated band insulator. Above Tc, slight hole (electron) doping introduces a striking dichotomy between electron and hole pockets: The electron (hole) pocket develops a pseudogap while the other becomes a nearly incipient band. We reveal that the superconductivity is driven by kinetic (potential) energy gain in the underdoped (overdoped) region. We also find a very short coherence length, for which we argue the relevance to multiorbital physics. Our Letter offers crucial insights into the superconductivity in the bilayer Hubbard model potentially relevant to La3O2O7.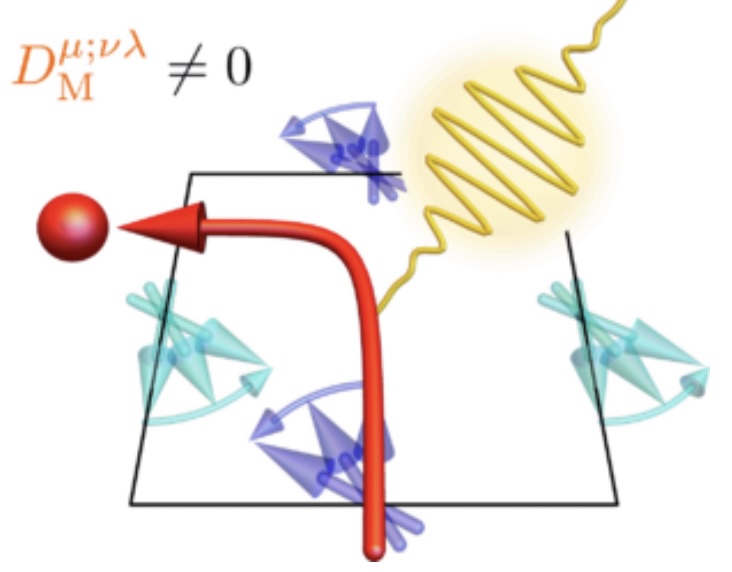 Parity-time-reversal symmetric (PT-symmetric) magnets have garnered much attention due to their spin-charge coupled dynamics enriched by parity-symmetry breaking. By real-time simulations, we study how localized spin dynamics can affect the nonlinear Hall effect in PT-symmetric magnets. To identify the leading-order term, we derive analytical expressions for the second-order optical response and classify the contributions by considering their transformation properties under PT symmetry. Notably, our results reveal that sizable contribution is attributed to the mixed dipole effect, which is analogous to the Berry curvature dipole term.
Parity-time-reversal symmetric (PT-symmetric) magnets have garnered much attention due to their spin-charge coupled dynamics enriched by parity-symmetry breaking. By real-time simulations, we study how localized spin dynamics can affect the nonlinear Hall effect in PT-symmetric magnets. To identify the leading-order term, we derive analytical expressions for the second-order optical response and classify the contributions by considering their transformation properties under PT symmetry. Notably, our results reveal that sizable contribution is attributed to the mixed dipole effect, which is analogous to the Berry curvature dipole term.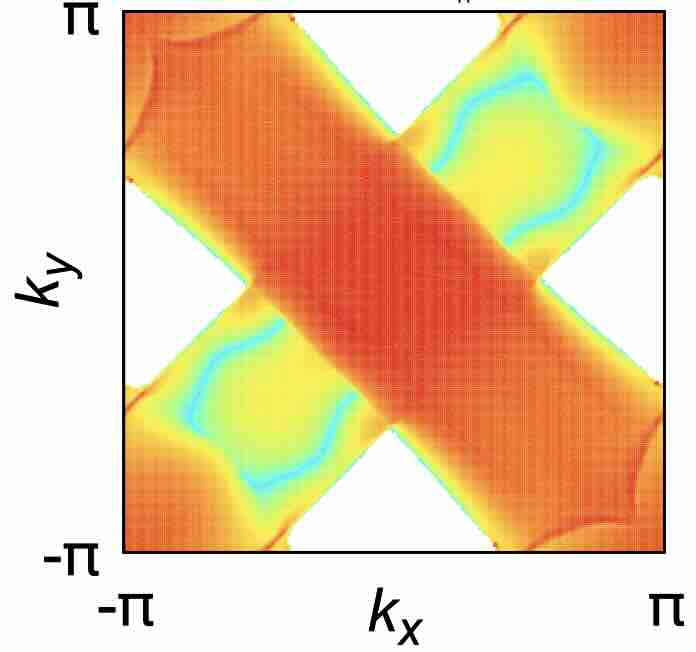 We investigate the functionality of the Cr-doped RuO2 as an electrode of the magnetic tunnel junction (MTJ), motivated by a recent experiment showing that Cr doping into rutile-type RuO2 is an effective tool to easily control its antiferromagnetic order and the resultant magnetotransport phenomena. We perform first-principles calculation of the tunnel magnetoresistance (TMR) effect in the MTJ based on the Cr-doped RuO2 electrodes. We find that a finite TMR effect appears in the MTJ originating from the momentum-dependent spin splitting in the electrodes, which suggests that RuO2 with Cr doping will function effectively as the electrode of the MTJ. We also show that this TMR effect can be qualitatively captured by the local density of states inside the tunnel barrier.
We investigate the functionality of the Cr-doped RuO2 as an electrode of the magnetic tunnel junction (MTJ), motivated by a recent experiment showing that Cr doping into rutile-type RuO2 is an effective tool to easily control its antiferromagnetic order and the resultant magnetotransport phenomena. We perform first-principles calculation of the tunnel magnetoresistance (TMR) effect in the MTJ based on the Cr-doped RuO2 electrodes. We find that a finite TMR effect appears in the MTJ originating from the momentum-dependent spin splitting in the electrodes, which suggests that RuO2 with Cr doping will function effectively as the electrode of the MTJ. We also show that this TMR effect can be qualitatively captured by the local density of states inside the tunnel barrier.